

Мами. Терористки
Ледь не щокварталу вони влаштовують вуличні протести під урядовими будівлями. Постійно з’являються в новинах. Засипають листами та зверненнями скриньки очільників країни, а особливо — чиновників Міністерства охорони здоров’я. Ті ж позаочі називають їх терористками. І кожна нова команда у міністерстві має з ними конфлікт. Вони — це матері й батьки дітей, хворих на рідкісні генетичні хвороби, активісти громадської спілки «Орфанні захворювання України». Вже шість років вони ведуть щоденну війну за право пацієнтів отримувати лікування. Ми спробували розібратися, чому ухвалення будь-якого рішення їм доводиться буквально домагатись і чи цей конфлікт батьків і держави може колись вичерпатись.
***
Плече до плеча. Обидва входи до міністерської будівлі загороджують із десяток охоронців у чорному. Обличчя закриті масками — видно самі очі. Скільки ж протестних акцій вони бачили? Навіть тепер, коли в країні уже восьмий тиждень триває карантин і кожен мав би залишатися вдома, під вікнами Міністерства охорони здоров’я галасують пікетувальники. Точніше, пікетувальниці.
Журналістів із камерами й мікрофонами вдвічі більше за самих активісток. Через епідемічні обмеження збиратися великими групами заборонено, але протестувальниці знайшли вихід — замість пацієнтів, чиї права відстоюють, вивели на пікет пів сотні картонних манекенів. Дочекатися закінчення карантину, кажуть, не могли.
— Якщо ви не почуєте нас зараз, — здіймаючи очі до міністерських вікон, говорить у мегафон одна з активісток, — наступного разу нам доведеться прийти сюди не з пацієнтами, а зі свідоцтвами про їхню смерть.
Це Таня Кулеша — головний двигун організації, та, чиє ім’я викликає роздратування у людей у міністерських кабінетах. Пізніше вона пояснить, що не перебільшує. Частина пацієнтів, які мають рідкісні генетичні захворювання, справді можуть померти, якщо матимуть перерву в лікуванні або вживатимуть не ті препарати, які їм призначив лікар. А саме до того, каже, йде.


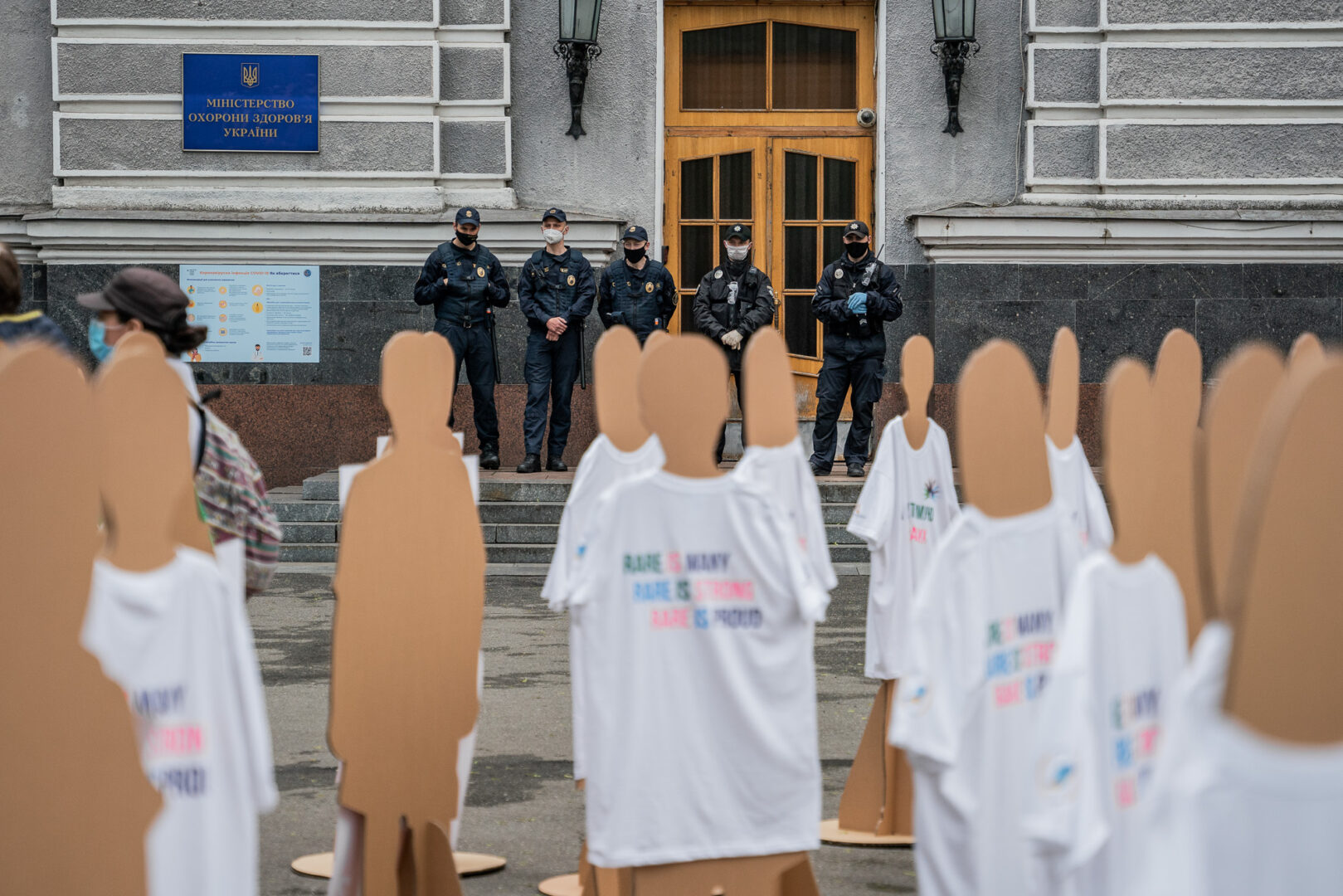
— Холодно шо капєц, — застібаючи літній плащ, говорить високий хлопець років 30. Він чи не єдиний чоловік зі сторони організаторів.
— Нам не звикати, — сміється його колега. — А що, — питає вона, — плакати з манекенів знімаємо зараз чи вже в офісі?
— Знімаємо тут. І все разом відвеземо в офіс. У нас за шість років там уже цілий склад.
***
Відлік організації ведуть відтоді, як почалася робота над законом про орфанні захворювання — його ухвалили у квітні 2014-го. Закон чітко визначив: держава зобов’язується лікувати пацієнтів із рідкісними захворюваннями й виділятиме на це гроші. Та ейфорія від появи документа минула, щойно стало зрозуміло, що коштів у держбюджеті немає, а які саме хвороби вважати рідкісними — не ясно, скільки таких пацієнтів — ніхто достеменно не знає, бо немає реєстру хворих.
Тоді троє мам вирішили, що мусять брати все у свої руки. Одна вже встигла поховати півторарічну доньку, яка померла від невідомої хвороби, і шукала способів не втратити ще й старшого сина, якому діагностували рідкісну недугу — мукополісахаридоз. 10-річний син іншої страждав на генетичну хворобу шкіри, бульозний епідермоліз. Дитину третьої забрала легенева гіпертензія — дівчина-підліток помирала за зачиненими дверима квартири, а мати не могла потрапити всередину, щоб допомогти.
Після шести років громадська спілка «Орфанні захворювання України» — це вже не просто волонтерський проєкт кількох мам, а велика організація, що об’єднує спільноти пацієнтів по всій країні і входить до європейської мережі Eurordis. Вони не єдині, хто захищає в Україні права орфанних пацієнтів, але точно найпомітніші.
***
— Імовірність, що у здорових батьків народиться дитина з рідкісним діагнозом, становить 25 %, — говорить лікарка-генетикиня Наталія Самоненко. Вона працює в Національній дитячій лікарні «Охматдит» і є спеціалісткою з лікування та діагностики пацієнтів із патологіями обміну речовин та інших процесів, зумовлених спадковою схильністю.
— Ми всі є носіями зламаних генів, — каже лікарка. — Ніхто не застрахований від того, що у вас і вашої половинки ці зламані гени не співпадуть, призвівши до хвороби дитини. Крім того, трапляються також і спонтанні мутації. Це не означає, що кожному варто боятися народжувати, але, водночас, до вагітності треба підходити обдумано, плануючи її та консультуючись із генетиком.
Легенева гіпертензія не дає дихати та нищить серце. Бульозний епідермоліз безжально ранить шкіру. Недосконалий остеогенез кришить кістки. Спінальна м’язова атрофія паралізує м’язи. Такі захворювання трапляються рідко — рідше ніж один випадок на 2000 населення. Але різновидів орфанних хвороб багато, понад 6 тисяч. Тож у загальному людей з орфанними діагнозами мільйони — тільки в Європейському Союзі понад 30 мільйонів. В Україні точних цифр ніхто не знає — держава має на обліку лише 5 тисяч хворих, тоді як світова статистика дозволяє припустити: якщо йдеться про 5 % населення країни, то недужих може бути близько двох мільйонів.
— Діагностувати орфанні захворювання в Україні можна, але не всі люди мають рівний доступ до діагностики. Часто для того, щоб потрапити до лікаря, який спеціалізується на тій чи іншій хворобі, людині треба їхати в інший регіон. Потім здавати аналізи, частину яких в Україні не роблять або роблять лише платно. Щоб підтвердити діагноз, українські лабораторії часто надсилають матеріали для скринінгу за кордон, що відповідно коштує, — пояснює лікарка.
Схожа ситуація з лікуванням. Препарати, які можуть допомогти орфанним пацієнтам, — дорогі. Інколи йдеться про сотні тисяч, а то й мільйони гривень — ліки виготовляють за новітніми біотехнологіями та для обмеженої кількості пацієнтів. При масовому виробництві популярних препаратів фармакологічні фірми заробляють і за низьких цін, але якщо препарат потрібен лише кільком сотням пацієнтів у всьому світі, витрати на його розробку й виробництво просто не можуть окупитися. Тому так важливо, щоб держава брала закупівлю ліків на себе.
***
Орфанні хвороби невиліковні. Та розвиток частини з них можна втримати: що раніше хвороба буде виявлена і що скоріше розпочнеться терапія, то більше шансів на повноцінне життя матиме людина. Як-от семирічна Пелагія. Вона — третя й дуже бажана в родині дитина. Утім, через три місяці після народження її батьків шокувала новина від лікарів: дівчинка має вроджену хворобу, муковісцидоз. Це важке спадкове захворювання, яке вражає частини залоз і підвищує в’язкість секретів, що призводить до ураження легень, підшлункової та кишківника, рідше — печінки та нирок. Щоб жити, дівчинка має постійно робити інгаляції та вживати додаткові ферменти.
— Простими словами, її організм просто не здатний виводити мокроту, яка збирається, в нашому випадку, у легенях. Якщо її не викашлювати (що у здорових людей відбувається природно), в легенях накопичуються бактерії. Тому нам із Пелагією доводиться виводити мокроту через інгаляції спеціальними препаратами, — розповідає мама дівчинки Катерина.
Сім інгаляцій щодня. Кожна по 20-30 хвилин. Крім того, Пелагія змушена ще й ковтати десятки пігулок — хвороба зачіпає і травну систему: в організмі майже не засвоюються ні білки, ні жири, ні вуглеводи. Таблетки, які вона мусить пити щодня, ледве поміщаються у маленьку долоню. Цілком дорослий виклик для семирічної дівчинки.
Та це не заважає Пелагії лишатися дитиною. Бавиться, інколи дуркує, але здебільшого наполегливо рухається до мети — стати гімнасткою. Щоденні тренування, а крім того — заняття танцями та уроки у школі моделей.

— Зовні по ній не можна сказати, що вона чимось відрізняється від інших. Активна, життєрадісна і дуже наполеглива дитина, — говорить мама. — В європейських країнах, де муковісцидоз лікують строго за протоколами, а пацієнтів забезпечують якісними препаратами, люди з таким діагнозом живуть до 40-50 років. У нашій країні, на жаль, статистика інакша.
***
Ірина сама лікарка. Утім, навіть власний фах не пом’якшив новину про те, що обоє її дітей мають рідкісні хвороби. Першим діагноз поставили синові. Сергію було 10, коли через часті носові кровотечі, нестерпні головні болі, відставання у вазі та суттєво збільшену селезінку довелося робити комплексну діагностику й шукати причину. Після трьох місяців досліджень її таки визначили — зламаний ген. У хлопця виявили хворобу Гоше — генетичне захворювання, що спричиняє дефіцит ферменту, який розщеплює і виводить з організму жири. Зрештою вони відкладаються в клітинах різних органів, найчастіше — у печінці й селезінці, перетворюючи здорову тканину на рубцеву.
Заради власного спокою батьки зробили таке дослідження і Сергієвій молодшій сестрі Серафимі, хоч тоді вона не мала жодних симптомів. Рідкісний діагноз підтвердився і у трирічної дівчинки. «Ніколи в житті не плакала так, як тоді», — пригадує Ірина.
Відтоді минуло чотири роки. Після початку лікування — що два тижні дітям крапають спеціальний препарат — розвиток хвороби вдалося призупинити. А збільшена селезінка у хлопця помітно зменшилась. Та ситуація все одно лишається критичною — будь-які перерви у лікуванні небезпечні. Минулого року через затримки з поставками й нестачу ліків Сергієві зменшили дозу препарату — і хвороба знову почала прогресувати. Крім того, в обох дітей завжди є небезпека внутрішніх кровотеч. Тому вони не можуть займатися спортом і мусять бути обережними.
— Сергій доволі інтровертний підліток, — розповідає мама. — Має кількох друзів, але здебільшого проводить час за читанням книжок. Серафимі вже сім, вона мріє то про кота, то про собаку, але ми не можемо завести домашню тварину, бо в мене алергія. Вона ще мріяла про бальні танці. Ми навіть почали ходити на заняття, але змушені були полишити. Хвороба пригнічує імунітет, тому Сіма часто застуджується.

Для пацієнтів із хворобою Гоше, як, зрештою, і для інших орфанних пацієнтів, важливо не тільки не переривати лікування, а й підібрати його правильно. Зробити це можуть лікарі, спираючись на досвід, протоколи лікування та спостереження за індивідуальною реакцією організму. Тільки-от рішення про те, які саме препарати будуть закуплені за гроші держави, ухвалюють не вони. Пів року тому Міністерство охорони здоров’я вирішило об’єднати три різні препарати для лікування хвороби Гоше в один лот і закупити найдешевший. Це зрозуміло з точки зору економії бюджету, але не дуже — з погляду самих пацієнтів та лікарів.
— Якщо всіх пацієнтів перевести на лікування одним препаратом, то в частини з них можуть виникнути небезпечні побічні реакції. І ті, кому препарат не підійде, залишаться без лікування взагалі. Наше міністерство говорить, що така практика діє у країнах ЄС: Німеччині, Польщі, Чехії, Хорватії. Але ми перевірили інформацію на сайтах міністерств охорони здоров’я та інших відповідних органів цих країн і проконсультувалися з представниками пацієнтських асоціацій. Виявилося, що ні в Чехії, ні в Хорватії, ні в Німеччині, ні в Польщі до терапії хвороби Гоше такого підходу не застосовують, а препарати призначають індивідуально з огляду на стан здоров’я пацієнта, — розповідає Олена Мартиненко, голова Всеукраїнського об’єднання інвалідів — хворих на хворобу Гоше.
Ситуація зі списком ліків, які держава купує для пацієнтів за бюджетні кошти, і вивела активісток на черговий пікет — навіть попри карантин.
***
— Перелік ліків, так звані номенклатури, складає «постійна робоча група». По суті, це чиновники, які до лікування пацієнтів стосунку не мають. Є ще експертні групи, куди входять лікарі, але їхні висновки ігнорують, — пояснює на камери голова орфанної спілки Таня Кулеша, яку буквально обступили журналісти. Знімають так, щоб у кадр потрапляли картонні манекени, що стоять за спиною активістки. — Ми вже давно боремося за те, щоб цей орган діяв прозоро, а засідання були відкритими для лікарів та пацієнтських організацій.





Поки два десятки журналістів та кілька учасниць акції чекають на реакцію міністерства, Таня нарікає:
— З кожною новою командою, яка приходить у міністерство, маємо один і той самий сценарій. Ми приходимо сюди й вимагаємо, щоби нас почули. Нам обіцяють, що так і буде, говорять, що всі помилки — це робота попередників, просять дати їм трохи часу розібратися. А тоді нас знову починають ігнорувати до наступної зміни команди.
— Бо вони шантажують державу, — пояснює логіку дій міністерства один із колишніх менеджерів відомства, який погодився спілкуватись на умовах анонімності. — Об’єктивно лікування орфанних хворих дуже дороге і Україна собі цього дозволити не може. Плюс є інші напрямки, які треба розвивати і де значно більша кількість людей отримає допомогу за ті самі кошти.
Статистика на його боці: минулоріч на закупівлю ліків із держбюджету виділили 6,6 мільярда гривень, із яких, наприклад, на напрям дорослої онкології було скеровано 1,3 мільярда, і майже стільки ж — близько 1 мільярда — на орфанні хвороби. Тільки-от хворих на рак в Україні сотні тисяч (щороку близько 140 тисяч українців уперше дізнаються про такий діагноз), а орфанних хворих, яких держава узяла на облік, всього близько 5 тисяч. Але для матерів хворих дітей це не аргумент.
— Якщо б вас поставили перед вибором: боротися, попри нерозуміння інших, чи відступити й дати померти власній дитині? Ви б дивилися, як помирає ваш син чи донька, і думали би про те, що хтось інший тепер має шанс жити? — риторично запитує Таня Кулеша.
Коли її власному синові було 9, вона стояла перед таким вибором. Лікарі не давали хлопцеві шансів («лікування надто дороге, змиріться і народіть собі ще»), а вона дала. Почала шукати ліки й вимагати їхньої реєстрації та закупівлі. Сьогодні хлопцеві 21, він одружений і має власну справу — відкрив фірму з прокату повітряних куль, аби здійснювати мрії інших.
Боротьба цих жінок нагадує мені самовіддану й трагічну працю доктора Ріє, героя Альбера Камю, який навпомацки лікував хворих на чуму, знаючи, що всіх не порятує і що його робота — це нескінченна поразка. Але жодного разу не поставив свою роль під сумнів. Бо хіба можна не рятувати життя, якщо ти в силах спробувати?
***



Охоронці у чорному розступаються, і до пікетувальниць та журналістів виходять двоє людей з міністерства. На камери пояснюють: через епідемію COVID-19 і зміну команди робоча група не збиралась уже два місяці, в доцільності ухвалених до того рішень треба розбиратись і вони обов’язково це зроблять, дайте тільки час. «Упевнена, що спільно ми зможемо напрацювати рішення», — говорить заступниця міністра і майже одразу зникає за спинами охоронців.
Таке тут чули вже не раз.
Фотографії дітей із Василем Вірастюком та Іларією — Миколи Заварики.
Дочитали до кінця! Що далі?
Далі — невеличке прохання. Будувати медіа в Україні — справа нелегка. Вона вимагає особливого досвіду, знань і ресурсів. А літературний репортаж — це ще й один із найдорожчих жанрів журналістики. Тому ми потребуємо вашої підтримки.
У нас немає інвесторів чи «дружніх політиків» — ми завжди були незалежними. Єдина залежність, яку хотілося б мати — залежність від освічених і небайдужих читачів. Запрошуємо вас приєднатися до нашої Спільноти.
Репортажі801
Більше





Світ96
Більше

